The application of next generation sequencing to the diagnosis of inherited platelet disorder
What are inherited platelet disorders?
Platelets are small anucleate cells, produced in the bone marrow, which play a major role in blood clotting. Normally there are between 150,000 - 450,000 platelets/uL of blood. Patients with a low platelet count, defined as less than 50,000 platelets/uL of blood, are described as having thrombocytopenia and this may be a sign that the patient has an inherited platelet disorder.
The inherited platelet disorders (IPDs) are a large group of individually rare diseases characterised by one or more of the following: thrombocytopenia, altered platelet shape, size, structure and/or function. The main symptom of the IPDs is mucocutaneous bleeding of varying severity, ranging from easy bruising to severe bleeding after injury or surgery. Worldwide, the estimated frequency of the IPDs is around 1 in 10,0001 but this is likely to be an underestimate as many affected individuals have very mild or even no bleeding symptoms and often only come to medical attention following an incidental finding of thrombocytopenia during routine blood testing. Although severe bleeding is not an issue for the vast majority of patients, a number of the IPDs are associated with serious extra-haematological symptoms such as pre-senile cataracts, early onset sensorineural deafness, renal failure and an increased risk of developing cancer, especially leukaemia, so identifying those at risk is important for their future management and quality of life.
How are the IPDs diagnosed?
Currently diagnosis is based on bleeding history, platelet count and platelet function tests. However, these are labour intensive and not very well standardised between laboratories. In fact studies have shown that they are unable to detect a platelet defect in around half of the patients’ referred2.
Genetic analysis offers the potential of a quick and definitive diagnosis and clinicians are increasingly requesting this as a first line test if they suspect an IPD. However, deciding which gene to analyse can be difficult - platelet development is a complex, multistage process and involves the expression of many genes, and mutations in any one of these could potentially give rise to an IPD (Table 1). So far mutations have been found in more than 50 genes in association with platelet disorders.
In Viapath’s Molecular Haemostasis laboratory the targeted analysis of 15 genes by PCR and Sanger sequencing is currently offered. However as several genes may need to be analysed, diagnosis can be time consuming and costly. Next generation sequencing technologies offer a rapid method for determining the causative variant in affected individuals as they permit the analysis of multiple genes simultaneously. Thus a pilot study was carried out to investigate the validity of this approach.
Development of a new testing regime for IPD
A targeted exome sequencing was used to analyse a panel of 60 genes in 8 of our patients who had been investigated previously and in whom no putative pathogenic variant had been identified and the results are presented in Table 1.
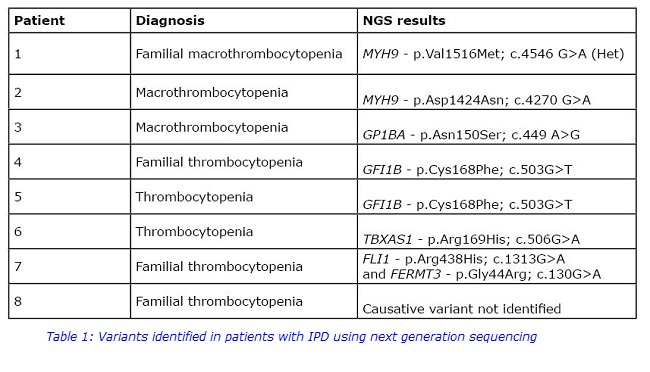
Putative pathogenic variants were identified in “platelet genes” in 7 of our 8 patients, five of which were clearly causative.
Patients 1 and 2 had a variant in MYH9, both of which have been reported in association with thrombocytopenia3. The MYH9 gene codes for the heavy chain of non-muscle myosin IIA, a cytoskeletal protein which is involved in several processes including cell motility and maintenance of cell shape. Some variants in MYH9 have been associated with early onset deafness and/or cataracts but these were not detected in this study.
Patient 3 was found to have a heterozygous variant in GP1BA. This gene codes for part of the GP1b-IX-V complex which is required for megakaryocyte maturation and normal platelet morphogenesis and GP1BA variants have been reported in autosomal dominant Bernard-Soulier Syndrome4.
Two unrelated patients, 4 and 5, were found to have a heterozygous variant in GFI1B, which has been reported previously in patients with mild to moderate thrombocytopenia5. Variants in GFI1B, which encodes a transcription factor, have been associated with an increased risk of developing leukaemia and other cancers6 so the patients will be informed and monitored.
Putative pathogenic variants were also identified in platelet-specific genes in 2 other patients which are likely to be responsible for their thrombocytopenia. A novel heterozygous mutation in TBXAS1 was identified in patient 6. Variants in this gene, which codes for thromboxane synthase, have been reported in autosomal dominant bleeding disorder, platelet type 147. However affected individuals usually present with significant bleeding, defective platelet aggregation and normal platelet count, which suggests that there may be other moderating factors contributing to the milder phenotype in this patient.
In patient 7 heterozygous variants in 2 genes, FLI1 and FERMT3, were identified. The transcription factor FLI1 is involved in regulating gene expression during platelet production and variants have been reported in association with mild thrombocytopenia, reduced dense granule secretion and symptoms such as eczema and alopecia8. A recent study found that FLI1 variants are common in patients with thrombocytopenia9. FERMT3 encodes kindlin which has been shown to be involved in integrin activation and inside-out signalling. To date only a few pathogenic variants have been reported in this gene, and all of them are associated with Leukocyte adhesion deficiency-3 which is characterised by immune deficiency and Glanzmann Thrombasthenia-like bleeding10. The phenotypic data on our patient was incomplete but they were reported to have only mild bleeding so analysis is ongoing to determine if these variants are truly causative.
Thus, using this new approach putative pathogenic variants were identified in all but 1 of our 8 patients which is a diagnostic hit rate of ~80%. These exciting results clearly demonstrate that next generation sequencing can provide a rapid molecular diagnosis in patients with IPDs even in the absence of detailed phenotypic data. This test is now being adopted as part of our diagnostic repertoire and it is anticipated that it will significantly improve patient treatment and prognosis.
For further information on this new test, please contact:
Molecular Haemostasis Laboratory
4th Floor North Wing
St Thomas Hospital
SE1 7EH
020 7188 2798
References
- Bolton-Maggs PHB et al., A review of inherited platelet disorders with guidelines for their management on behalf of the UKHCDO. Br J Haematol 2006; 135: 603–33.
- Watson SP et al., Genotyping and phenotyping of platelet function disorders. J Thromb Haemost 2013; 11 (Suppl. 1): 351–63.
- Balduini et al., Recent advances in the understanding and management of MYH9-related inherited thrombocytopenia. Br J Haematol 2011; 154: 161-174.
- Savoia A et al., Autosomal dominant macrothrombocytopenia in Italy is most frequently a type of heterozygous Bernard-Soulier syndrome. Blood 2001; 97: 1330-1335.
- Rabbolini DJ et al., The GFI1B, c.503G>T mutation in the first zinc finger domain predicts a milder bleeding phenotype than the GFI1B c.880-881insC mutation. J Thromb Haemost 2015; 13 (Suppl S2):659.
- Anguita E et al., Transcription Factor GFI1B in Health and Disease. Frontiers in Oncology 2017; 7: 1-7.
- Defreyn G et al., Familial bleeding tendency with partial platelet thromboxane synthetase deficiency: reorientation of cyclic endoperoxide metabolism. Brit J Haematol 1981; 49: 29-41.
- Saultier P et al., Macrothrombocytopenia and dense granule deficiency associated with FLI1 variants: ultrastructural and pathogenic features. Haematologica 2017; doi:10.3324/haematol.2016.153577
- Stockley et al., Enrichment of FLI1 and RUNX1 mutations in families with excessive bleeding and platelet dense granule secretion defects. Blood 2013; 122: 4090-4093.
- Kuijpers TW et al., LAD-1/variant syndrome is caused by mutations in FERMT3. Blood 2009; 113: 4740-4746.