New methodology for diagnosing Huntington Disease
What is Huntington disease?
Huntington disease (HD) is an inherited neurodegenerative disorder. Typically it is late onset with symptoms first appearing between 35-50 years. There is a more severe form of the disease, known as juvenile HD, which has a much earlier onset. The disease is progressive and symptoms vary but can include involuntary muscle movements, emotional problems, speech loss and dementia. HD is usually fatal about 15 to 20 years after symptoms start.
Huntington disease is caused by mutations in the HTT gene, which provides instructions for the production of a protein called huntingtin. The function of this protein is still unknown but it appears to play an important role in the neurons of the brain. The HTT mutation involves a region of DNA that consists of repeated cytosine, adenine, and guanine (CAG) nucleotides. This is known as a trinucleotide repeat. Normally, the CAG trinucleotide is repeated 6 to 35 times within the gene. However, in people with Huntington disease, the CAG segment is repeated more than 35 times. This leads to the production of an abnormally long version of the huntingtin protein which eventually leads to the death of neurons in certain areas of the brain. The number of CAG repeats relates to the severity of the disease; cases of juvenile HD are associated with 60+ repeats.(1,2)
Current diagnostic testing for Huntington disease
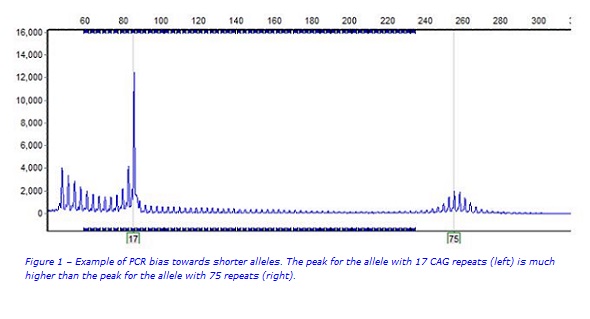
Current testing for Huntington disease involves amplification of the HTT CAG trinucleotide repeat region using a fluorescent polymerase chain reaction (PCR) assay. The fluorescent PCR products are accurately sized on a capillary sequencer with an internal size standard that allows the determination of the CAG repeat count for each of the two copies (alleles) of the HTT gene in an individual. However an issue with this approach is that PCR preferentially amplifies smaller fragments over larger fragments. This can result in ‘drop out’ of very large alleles (e.g. some of those responsible for juvenile HD) meaning they are not detected (Figure 1).(3)
Can diagnostic testing for Huntington disease be improved?
Viapath has recently introduced Oxford Nanopore long read sequencing technology into its Genetics Laboratory. This technology allows us to rapidly sequence long stretches of DNA using a small palm-sized device. Unlike short read ‘next generation’ sequencing; long read sequencing provides the ability to sequence the entire HTT CAG repeat region in a single read. It was therefore decided to investigate whether this technology could match the performance of traditional methods for diagnosing HD, and improve the ability to detect larger alleles.
Methodology
Forty-eight samples previously tested by PCR with CAG repeat counts ranging from 15 to 101 were selected. Two kilobase long-range PCR products covering the HTT CAG repeat region were generated. Libraries were constructed and sequenced on an Oxford Nanopore MinION. The error rate of Oxford Nanopore sequencing made data analysis challenging. We therefore ran all data through an algorithm called repeatHMM (https://github.com/WGLab/RepeatHMM) which applies a Hidden Markov statistical model to estimate the repeat count.(4)
Results
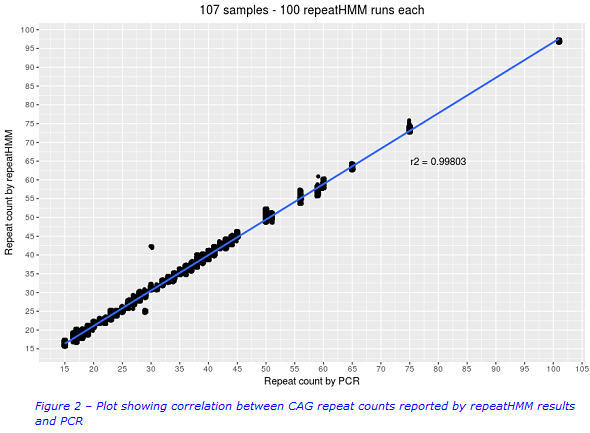
All of the alleles (15 – 101 repeats) were successfully detected by repeatHMM. A high correlation between the repeatHMM repeat counts and the PCR repeat counts was observed (Figure 2). Results from repeatHMM and PCR generally fell into the same HD classification categories. Best practice guidelines state that acceptable error limits are +/-1 repeat for alleles that are <42 repeats in length. Repeat counts in the intermediate and reduced penetrance ranges (where accuracy is most critical) were generally within +/-1 repeat of the PCR result. It was observed that repeatHMM slightly overestimated results at the lower limits and underestimated results at the upper limits of the repeat sizes. It was also observed that some outliers required further investigation. However the initial results were very promising.
Is this test the way forward for Huntington disease?
In the first instance we are implementing Oxford Nanopore sequencing as a reflex test in juvenile HD cases where we have failed to detect a pathogenic allele. Based on the promising initial results, we are hopeful that, with a little more optimisation, this test will be used more widely. We are working with Oxford Nanopore Technologies and other laboratories to share expertise and resources, which is allowing the development of this and other exciting new Nanopore tests. We are enthusiastic about the breadth of opportunities this new cutting edge technology presents us with and are grateful to the Viapath Innovation Fund for funding this project.
For further information please contact:
Andrew Bond, Pre-registration Scientist (Bioinformatics):
andrew [dot] bond5 [at] nhs [dot] net
References
1. Bates, G. P. (2005). History of genetic disease: the molecular genetics of Huntington disease - a history. Nature Reviews. Genetics, 6(10), 766–773. https://doi.org/10.1038/nrg1686
2. Gonzalez-Alegre, P., & Afifi, A. K. (2006). Clinical characteristics of childhood-onset (Juvenile) Huntington disease: report of 12 patients and review of the literature. Journal of Child Neurology, 21(3), 223–229. https://doi.org/10.2310/7010.2006.00055
3. Jama, M., Millson, A., Miller, C. E., & Lyon, E. (2013). Triplet repeat primed pcr simplifies testing for huntington disease. The Journal of Molecular Diagnostics, 15(2), 255–262. https://doi.org/10.1016/j.jmoldx.2012.09.005
4. Liu, Q., Zhang, P., Wang, D., Gu, W., & Wang, K. (2017). Interrogating the “unsequenceable” genomic trinucleotide repeat disorders by long-read sequencing. Genome Medicine, 9(1), 65. https://doi.org/10.1186/s13073-017-0456-7